The mageck-vispr toolkit is used to QC, analyse, and visualize results from CRISPR/Cas9 screens. It includes the
The mageck workflow is implemented as a snakemake pipeline and runs automatically. Vispr on the other hand is essentially a web application that will run a temporary server on a compute node and the user will connect to it using a browser on his/her own computer through an ssh tunnel.
$MAGECK_VISPR_TEST_DATAAllocate an interactive session with sinteractive and use as shown below. See our tunneling page for more details on setting up ssh tunnels.
biowulf$ sinteractive --cpus-per-task=8 --mem=16g --tunnel
salloc.exe: Pending job allocation 31864544
salloc.exe: job 31864544 queued and waiting for resources
salloc.exe: job 31864544 has been allocated resources
salloc.exe: Granted job allocation 31864544
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn2692 are ready for job
srun: error: x11: no local DISPLAY defined, skipping
Created 1 generic SSH tunnel(s) from this compute node to
biowulf for your use at port numbers defined
in the $PORTn ($PORT1, ...) environment variables.
Please create a SSH tunnel from your workstation to these ports on biowulf.
On Linux/MacOS, open a terminal and run:
ssh -L 46763:localhost:46763 biowulf.nih.gov
For Windows instructions, see https://hpc.nih.gov/docs/tunneling
[user@cn3144]$ module load mageck-vispr
Copy test data and run mageck pipeline
[user@cn3144]$ cp -r ${MAGECK_VISPR_TEST_DATA}/esc-testdata .
[user@cn3144]$ cd esc-testdata
[user@cn3144]$ mageck-vispr init ./test_workflow --reads \
reads/ERR376998.subsample.fastq \
reads/ERR376999.subsample.fastq \
reads/ERR377000.subsample.fastq
[user@cn3144]$ tree test_workflow
test_workflow/
|-- [user 2.9K] config.yaml
|-- [user 7.3K] README.txt
`-- [user 5.8K] Snakefile
0 directories, 3 files
[user@cn3144]$ cd test_workflow
Before running the workflow it is necessary to edit the automatically generated config file. The generated file contains many comments. Here is the edited file with the comments stripped for readability:
library: ../yusa_library.csv
species: mus_musculus
assembly: mm10
targets:
genes: true
sgrnas:
update-efficiency: false
trim-5: AUTO
len: AUTO
samples:
esc1:
- ../reads/ERR376999.subsample.fastq
esc2:
- ../reads/ERR377000.subsample.fastq
plasmid:
- ../reads/ERR376998.subsample.fastq
experiments:
"ESC-MLE":
designmatrix: ../designmatrix.txt
# the following setting is required for 0.5.6
correct_cnv: false
Once the config file has been modified to reflect the experimental
design, run the pipeline. Note that snakemake is used to run this locally, not
by submitting tasks as cluster jobs. Note that the snakemake installation
used for mageck-vispr has been renamed to mageck-vispr-snakemake
to avoid interfering with the general use snakemake.
[user@cn3144]$ mageck-vispr-snakemake --cores=$SLURM_CPUS_PER_TASK
Next, start the vispr server for visualization. Note that sinteractive sets the
$PORT1 variable to the port selected for tunneling.
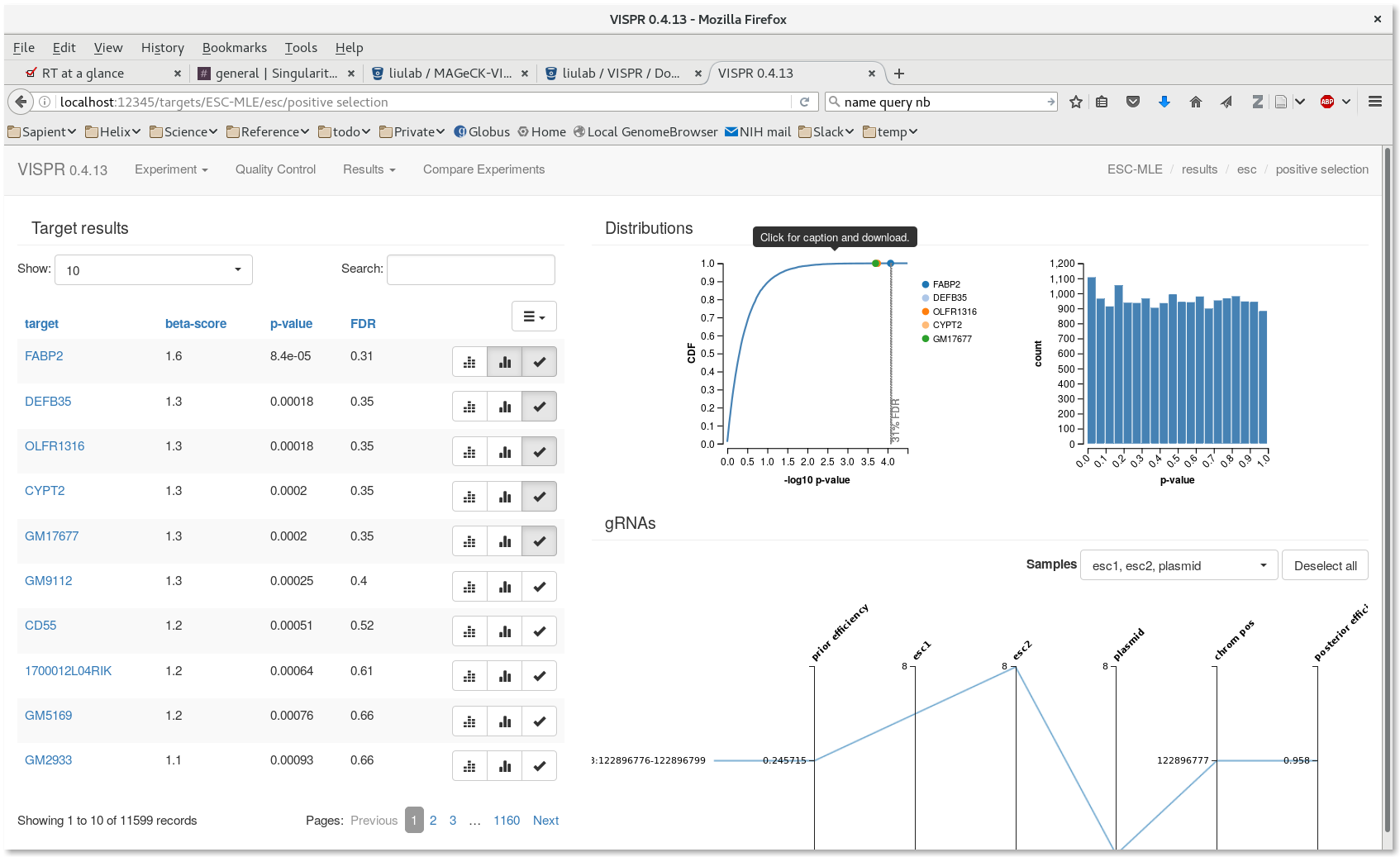
[user@cn3144]$ cd test_workflow [user@cn3144]$ vispr server --port $PORT1 --host=localhost results/ESC-MLE.vispr.yaml Loading data. Starting server. Open: go to localhost:39335 in your browser. Note: Safari and Internet Explorer are currently unsupported. Close: hit Ctrl-C in this terminal.
On your local workstation, create an ssh tunnel to biowulf as describe in our tunneling documentation.
Create a batch input file (e.g. mageck-vispr.sh), which uses the input file 'mageck-vispr.in'. For example:
Create a batch script for an existing config file similar to the following example:
#! /bin/bash # this file is mageck.batch module load mageck-vispr/0.5.4 || exit 1 cd /path/to/workdir mageck-vispr-snakemake --cores=$SLURM_CPUS_PER_TASK
Submit to the queue with sbatch:
biowulf$ sbatch --cpus-per-task=8 --mem=16g mageck.batch